USP chapter 〈1790〉 titled ‘Visual Inspection of Injections’, is the most efficient document that describes every single aspects which should be taken care while performing the validation of visual inspection process for the sterile injectables.
Overview
Regulatory guidelines have expectation that the sterile solutions should be free from any kind of intrinsic and extrinsic particles as it can have a significant impact on the human when injected into the blood streams.
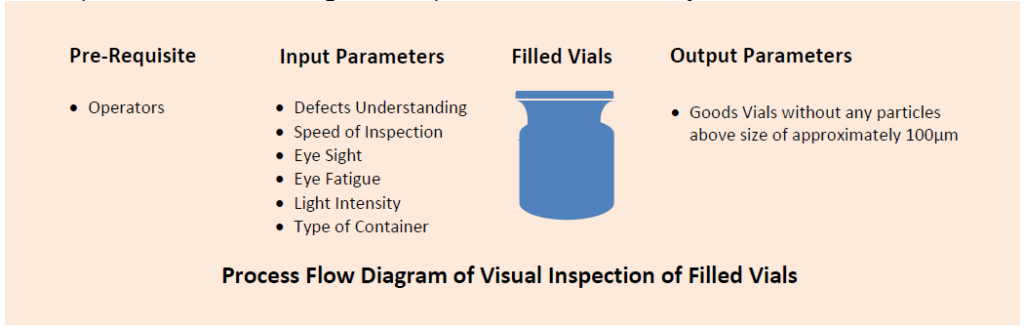
It is expected that the process of visual inspection of filled vials of the products is qualified and the inspectors are trained for the execution of the activity with real time defects to ensure 100% good vials after inspection.
Process of Visual inspection of injections is very important to minimize the introduction of unintended particles (process borne or environmental borne) to patients during the delivery of injectable in the form of medicine.
By process of Visual Inspection, compromised containers gets rejected, including those with cracks or incomplete seals, which pose a risk to the sterility of the product.
The desire to detect these defects, despite their very low frequency and the randomness of their occurrence, has resulted in the long standing expectation that each finished unit will be inspected (100% inspection). Although zero defects is the goal and this should drive continuous process improvement, zero defects is not a feasible specification for visible particles given current packaging components, processing capability and the probabilistic nature of the inspection process.
Human Inspection Process Capabilities

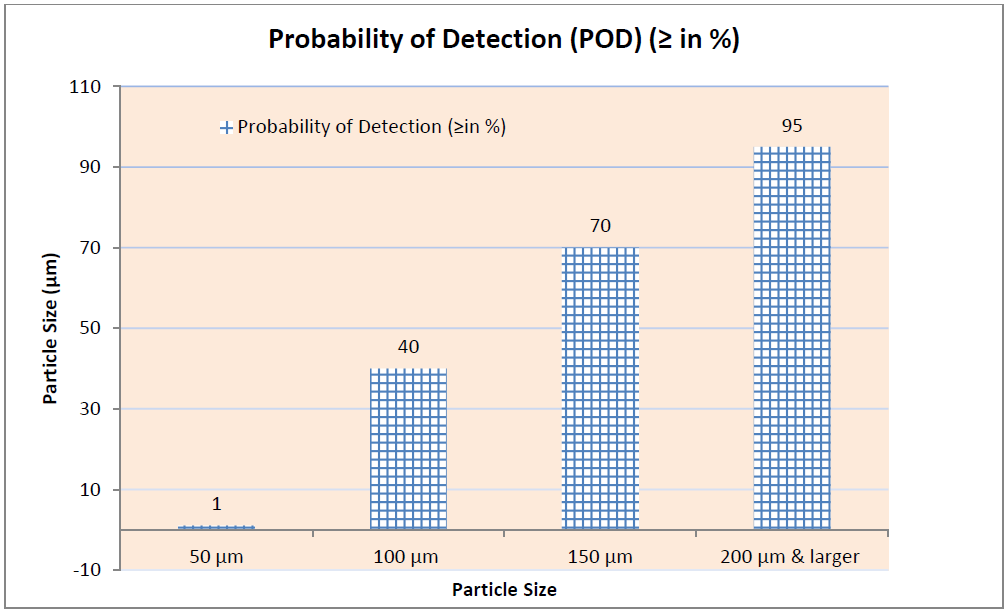
Note: This interpretation is made at 2,000 to 3,500 Lux for Clear Vial and 8,000 to 10,000 Lux for Amber Colored Vials.
Process of Qualification of Visual Inspection Process
DEGREE OF OPERATION AND PROCESS UNDERSTANDING
It is very important to understand the operation variability and the process. The more is the variability more complex goes the process. Hence is it important to have the broader spectrum when rationalizing the process and identifying the processes for the purpose of qualification.
Understanding the Origin of Defects or Contaminants
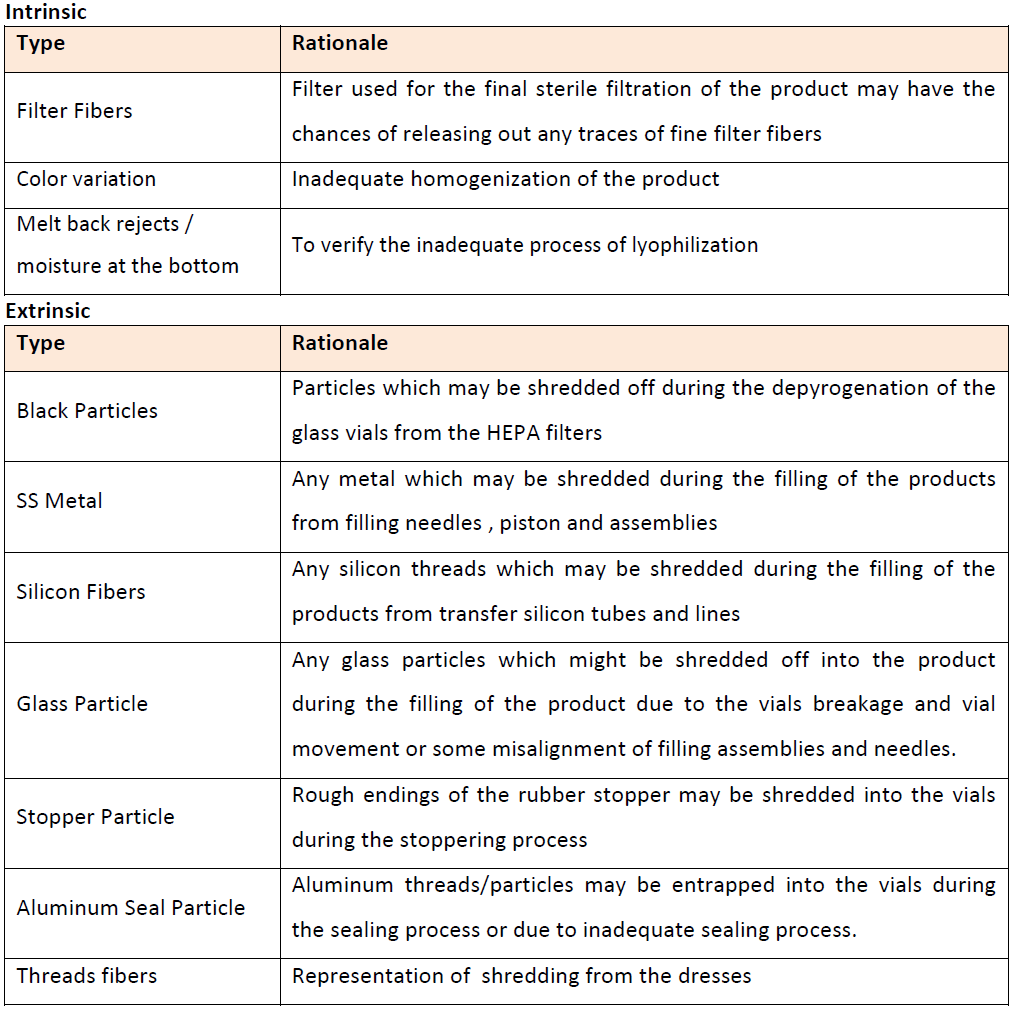
There are mainly three sources of defects or contamination of vials- Intrinsic, Extrinsic or Process Inherited
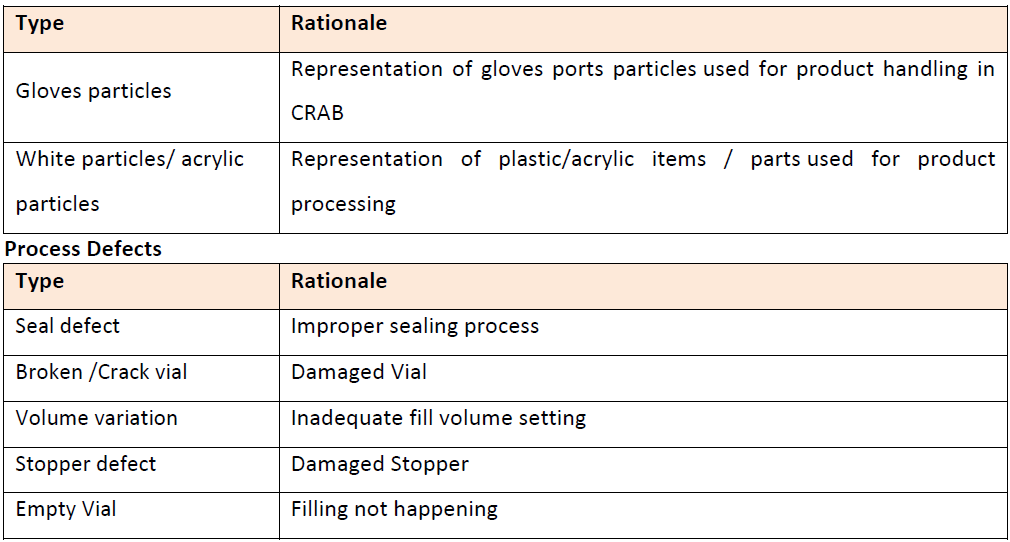
Determining the Container Size and Batch Size
Bracketing approach should be used for the determination of the batch size for the qualification kit. The vial with minimum and maximum should be considered as the containers for the purpose of qualification.
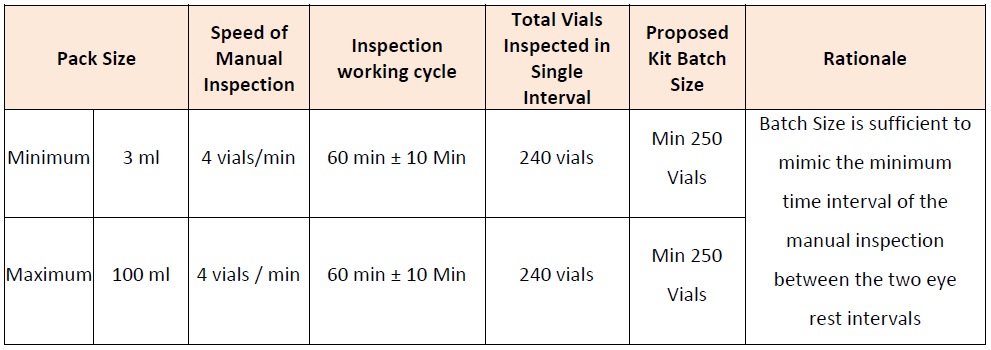
CREATION OF DEFECTS BASED UPON THE PRODUCT PROFILE AND CATEGORIZATION
Defects can be created artificially based upon the process requirements or can be considered from the ongoing process rejects. While creating the artificial rejects it is possible to go for the microscopic analysis of the size of the each particle which shall be introduced into the defect container.

PREPARATION OF QUALIFICATION KIT VIALS
Once defects are created and size estimation has been carried out, the good vials shall be added into the defects to complete the batch as per its lot size. Each vial should be properly numbered and identified with code. All the details of the defects should be maintained with codes.
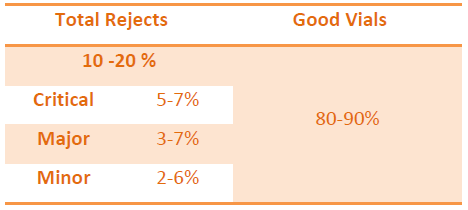
QUALIFICATION OF DEFECT KIT
Once the kit is prepared, next step is to qualify it before it is used for the qualification of the Visual Inspectors.
Inspection reject probability is calculated for the defect as follows where POD is termed as Probability of Detection:
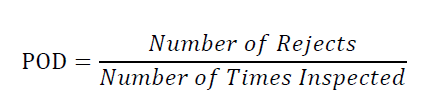
A manual, visual inspection with POD of ≥ 0.7 or 70 % is required to assign the container to the Reject Zone for subsequent calculation of the Reject Zone Efficiency (RZE).
Defect categorization should be uniform as per the available defects.
Acceptable Units are those which are showing POD (good vial of defected vial) of more than 70 % and same should be considered for the kit preparation.
Kit Qualification is important to rule out any container which shows variables results when inspected by different operators. Generally each container must undergo inspection by minimum 03 qualified inspectors for total of minimum 30 inspections to confirm the defect category.
DEFECT KIT MAINTENANCE
Should be ideally stored at the respective storage conditions for naturally occurring defects without leading to any unintended variations.
Expiry to be defined as 5 Years from the date of Preparation and may be reevaluated thereafter based upon the verification of defects.
QUALIFICATION OF THE VISUAL INSPECTORS
While qualifying visual inspectors, 03 runs of qualification should be performed to ensure the repeatability & consistency. Each visual inspector must undergo challenge test with qualified kit for the particular container as per the product characteristics.

TRAINING DEMONSTRATION
Demonstration of the inspection process shall be there for the new operators and following points should be the part of the training program.
- Vials Holding Technique
- Observation under white and black background
- Swirling and inverting of vials
- Verification of vial damage/defects
- Verification of height of meniscus
- Verification of seal/stopper defect etc. as examples
- Detection of defect vials from challenged lot
- Pictorial Representation & memorization of the Defects
- Scientific understanding on impact of the particles in human blood vessel system
ACCEPTANCE CRITERIA:
The acceptance criteria shall be as follows:
- Detection of 100% of critical defects
- Detection of ≥ 90% of major defects (For particulates)
- Detection of ≥ 80% of minor defects
- False Reject (goods vials) acceptance criteria are must be less than 5%.

Raman is a versatile experienced Bio-pharmaceutical professional with more than 17 year of experience in Sterile and Non-Sterile Formulations. Raman is working in different aspects of Sterile Validations and designing Pharmaceuitcals Quality Systems for the next century. He is a versatile and tech savvy professional who believe the Quality is the foundations of Growing Organizations







